Resources
Here are some resources you might find useful.
Scripts
Here are some scripts we built. Use and modify as you want.
Please, include a citation.
1. Filter-Sequences-by-Length

This script will filter out sequences from a fasta file that are not of a certain sequence length range. The Python script takes an input file containing sequences in fasta format, the output file name, the minimum length, and the maximum length. You can include a –visual flag at the end to have matplotlib create a histogram of the sequence lengths.
Example: python filter-by-length.py sequences.fasta sequences-filtered.fasta 200 250 –visual
You can download the script here
2. Gather Information on Sequences
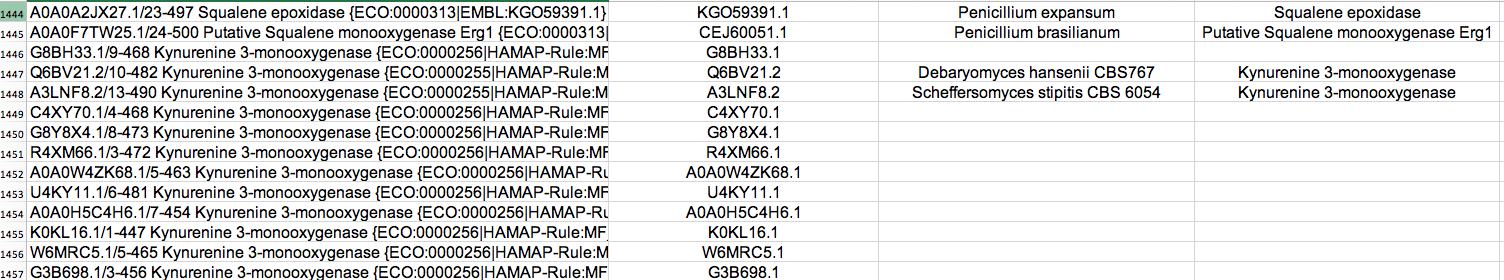 This script parses a file containing protein sequences in fasta format and downloads information
from EMBL or NCBI. The gathered information is then put into an Xcel
spreadsheet. The script may require some modifications for specific uses.
This script parses a file containing protein sequences in fasta format and downloads information
from EMBL or NCBI. The gathered information is then put into an Xcel
spreadsheet. The script may require some modifications for specific uses.
Example: python get_info.py sequences.fasta output.xlsx -emblebi -verbose
You can download the script here
3. Reorder Information on Sequences to correspond to the phylogenetic tree
This script reorders the information from an excel spreadsheet to match the order of a particular fasta file (one that may be ordered according to the phylogenetic tree). This script is useful for maintaining consistency between the sequence alignment, the meta-data on the sequences and the phylogenetic tree.
Example: python reorder_metadata.py sequences.fasta input.xlsx output.xlsx
You can download the script here
4. Group Entropy (GEnt)
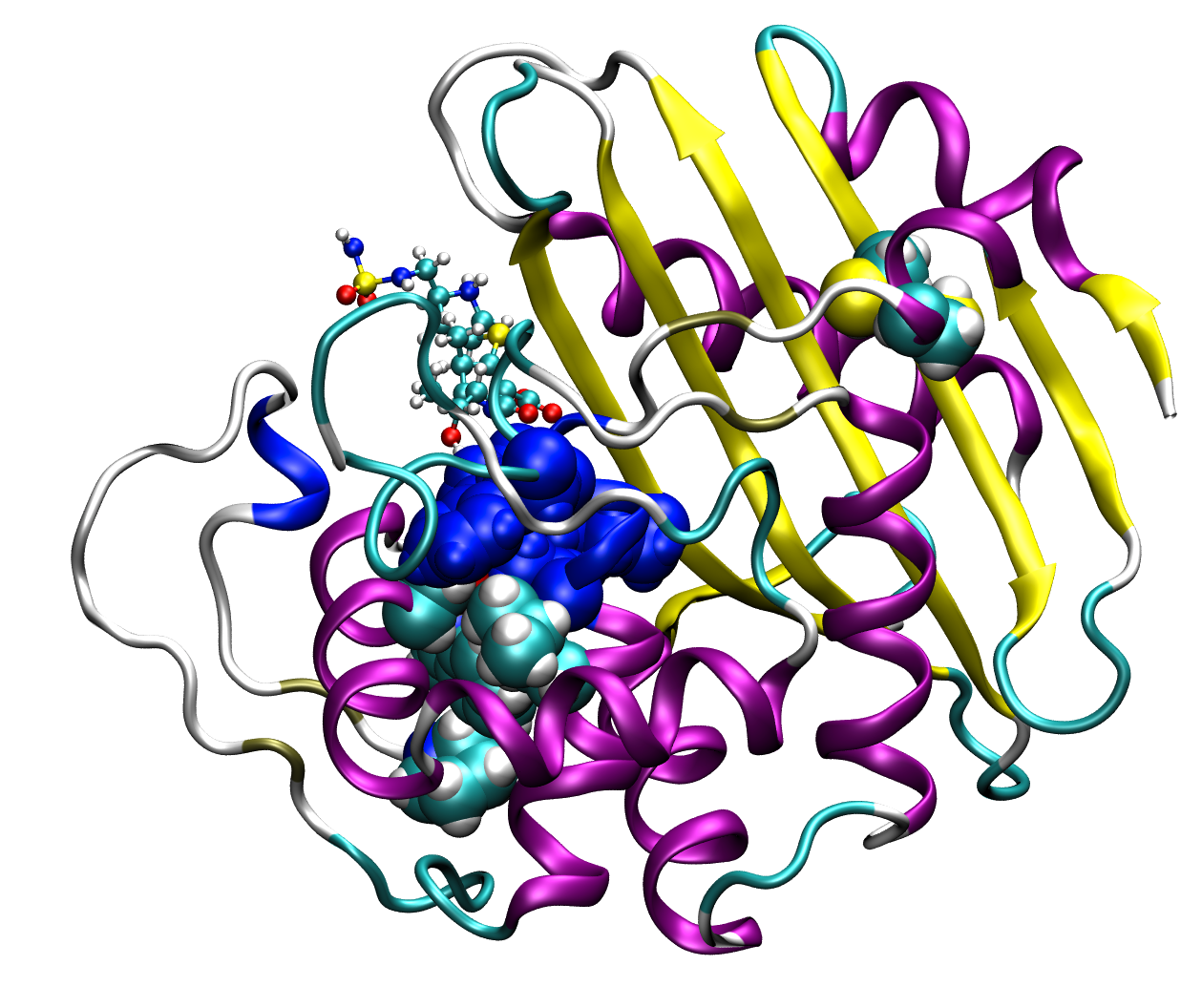
This program, written in Python, identifies residues within a clade (user must define what sequences are to be included in each clade) that are conserved within the clade but are different outside the clade and assigns a significance score to these sites. The following papers describe the method and applications of the method
- An Algorithm for Identification and Ranking of Family-Specific Residues, Applied to the ALDH3 family J. Hempel, J. Perozich, T. Wymore and H. B. Nicholas Jr. Chemico-Biological Interactions, 2003, 143-144:23-28. doi: 10.1016/S0009-2797(02)00165-5
- The class D Beta-lactamase Family: Residues Governing the Maintenance and Diversity of Function A. Szarecka, K. R. Lesnock, C. Ramirez-Mondragon, B. Y. Chen, H. B. Nicholas Jr. and T. Wymore* Protein Engineering, Design and Selection, 2011, 24:801-809.
- A Mechanism for Evolving Novel Plant Sesquiterpene Synthase Function T. Wymore*, B. Y. Chen, H. B. Nicholas Jr., A. J. Ropelewski and C. L. Brooks III Molecular Informatics, 2011, 30:896-906.
Author: Skylar Olson
| Example: python general_entropy.py input.fasta GrDel | PsMultiplier 4 RSequence XP3423 gapScore .90 |
You can download the script here